Abstract
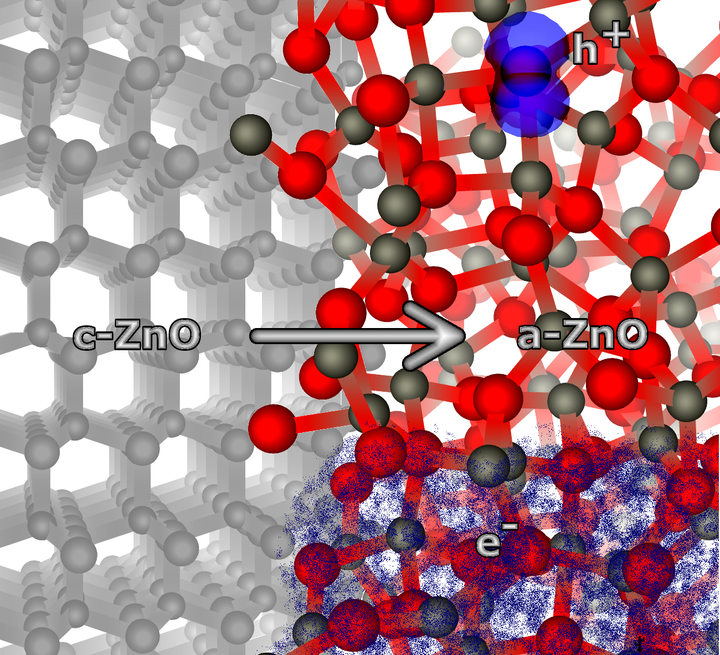
Recent advances in ultrafast liquid quenching and deposition of thin films on cold substrates make growing amorphous (a)-ZnO films increasingly feasible. We predict the electronic structure and electron and hole trapping properties of amorphous ZnO using Density Functional Theory (DFT) simulations with a hybrid density functional (h-DFT). An ensemble of fifty 324-atom structures is employed to obtain the distribution of structural and electronic properties of \mbox{a-ZnO}. The results demonstrate that electrons do not localize in \mbox{a-ZnO} but holes form deep localized states with average trapping energy of about \SI{0.9}{\electronvolt}. We also show that dispersion at the conduction band minimum (CBM) is not affected upon amorphization, suggesting that high electron mobility should be retained. An average value of a-ZnO band gap of \SI{3.36}{\electronvolt} is calculated with no states splitting into the band gap, which accounts for no substantial detrimental effect on the optical transparency upon amorphization. These findings may have important implications for future applications of \hbox{a-ZnO} as a transparent conductor and photo-catalyst.
David Mora-Fonz
Chemistry, Physics, Materials Science, Data Science, Decarbonisation, Climate Change
Spanish & English
I am a chemical engineer with a PhD and postdoc in Chemistry with likes for materials, energy, catalysis, electronics & data science